



内容
- 1 引言
- 2 聚合酶链式反应(PCR)
- 2.1 底漆二聚体形成
- 3 DNA测序
- 3.1 桑格(双脱氧)DNA测序
- 3.2 基于荧光二脱氧DNA测序
- 3.3 新一代测序
- 3.4 测序修饰的DNA
- 3.4.1 亚硫酸氢盐测序
- 4 DNA指纹
- 4.5 短串联重复序列
- 4.6 STR分析
- 4.7 荧光STR分析
- 4.8 亲子鉴定
- 5 单核苷酸多态性(SNP)
- 6 DNA诊断和突变检测
- 7 实时PCR
- 7.1 基于探测的实时PCR
- 7.2 的TaqMan方法
- 7.3 荧光共振能量转移(FRET)
- 7.4 分子信标
- 7.5 蝎子引物
- 8 其他诊断方法
- 8.1 DNA微阵列
- 9 荧光原位杂交(FISH)
介绍
21世纪初在我们的各种生物,最显着的例子是人类基因组测序的DNA序列的认识经历了一场革命。基因组DNA的分析使我们了解进化了很多,而DNA序列中隐藏的语言不同生物之间的关系,由基因控制的机制,对疾病的易感性。整个基因组的测序是尚未然而,常规技术中,和遗传分析的其它方法用于快速和有效地分析DNA样本。这些方法和技术,如DNA指纹图谱,也改变法医学。
DNA测序,基因和取证分析所有的已建立的方法依赖于使用标记的寡核苷酸和/或脱氧或双脱氧三磷酸核苷,和需要的DNA聚合步骤。这可以是聚合酶链反应(在遗传分析STR分析),单核苷酸延伸(微型测序SNP分析),或聚合和DNA链终止(的组合Sanger测序)。
聚合酶链反应(PCR)
聚合酶链反应(PCR)是在分子生物学,诊断学,法医学和分子遗传学广泛使用,以扩增一个特定区域(一个技术扩增子的DNA样品的)。PCR可以放大珍贵的DNA样本(如在犯罪现场)的几个分子产生大量的DNA,从50到长度超过25 000个碱基对。
在PCR中,两个短寡核苷酸(PCR引物)的设计,使得每个互补于在该区域的两个靶链之一的3'-端待扩增:两个PCR引物限定的扩增子。由引物结合模板的区域中的一系列的循环(扩增图1)。PCR需要的DNA聚合酶。而所有生物体含有DNA聚合酶,即在PCR中使用的聚合酶来自嗜热菌栖热菌属aquaticus。这Taq聚合酶是耐热的,这意味着高达95℃的温度下可在PCR中使用,低的条件DNA双链体的稳定性。
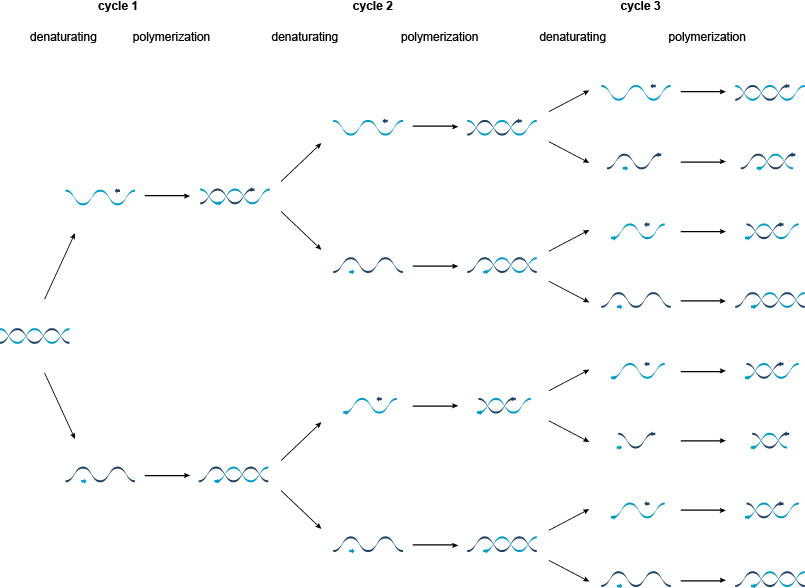
图1 | 的聚合酶链反应(PCR)
在第一周期中,双链靶被分离成通过加热两个单模板链至95℃。然后将其冷却至55℃,以允许合成的寡核苷酸引物退火到模板链与它们的3'端彼此相对。然后将温度升高到72℃,对于热稳定的Taq DNA聚合酶的活性最适温度。聚合酶使用脱氧核糖核苷三磷酸(dNTPs浓度),以引物沿着生产的DNA两个新的双链(模板的长度延伸。图2)。

图2 | PCR循环的聚合酶链式反应的单个周期。
PCR的第二周期是第一周期的重复,并且每个新合成的单链也可以作为引物退火和延伸的模板。聚合酶只能尽可能延长DNA作为第一引物的位点,产生特定长度的DNA双链体。在所有的后续周期的扩增产生的PCR产物由两个引物的位点特定的长度,而这些PCR产物很快多于原本的目标分子。在理论上,Ñ的PCR循环将产生2 Ñ PCR产物。
由凯利·穆利斯20世纪80年代发明的,聚合酶链反应已是穆利斯被授予了分子生物学,DNA诊断和法医学这样一个无处不在的影响诺贝尔奖。
引物二聚体的形成
不正确的扩增子在PCR中有时产生由于引物二聚体形成,其中,所述PCR引物杂交,和被放大的代替模板DNA(图3)。引物二聚体是最有可能以在PCR扩增开始时,当引物中存在的高浓度相对于模板。引物二聚体formatin可发生即使有几个错配的引物-二聚体双链体,其由结合于Taq聚合酶稳定。引物二聚体的形成是当几个PCR反应在同一个管(多重PCR)进行了一个特别的问题。

图3 | 引物二聚体形成的,而不是DNA模板扩增的形成与引物二聚体的扩增,可以对PCR产生不利影响。
在减少引物二聚体形成明显的步骤是,使它们具有低的自身互补来设计引物,但是这并不总是可能的,并且可能会导致引物二聚体的扩增甚至弱相互作用。“热启动”PCR技术已经发展到减轻引物二聚体的形成。在热启动PCR反应混合物最初缺少的重要组成部分,如Taq聚合酶,或Mg 2+(其中Taq聚合酶需要活性)。在第一周期开始长时间加热确保了所有的引物二聚体的变性,则该缺失的组件添加。PCR现在可以继续正常。该技术的主要缺点是污染的加入必须成分的,以打开反应管中的可能性。一个替代方法包括,关于加热解离的使用Taq聚合酶的非共价抑制剂(例如肽或抗体)。在另一方法中,PCR反应的一个重要组成部分是封闭在一个蜡丸,熔化在加热时,释放其内容。
DNA测序
所有这一切,因为在30年前由弗雷德·桑格在剑桥制定的方法是不可能的。桑格制定了关于他被授予他的第二个DNA测序(双脱氧法)的新方法诺贝尔文学奖,1980年。
桑格(双脱氧)DNA测序
桑格的DNA测序的双脱氧方法是常规地用于在实验室中的DNA测序的第一个方法。以下组件所需的Sanger测序:
- DNA模板进行测序
- 在5'端标记的与寡核苷酸引物32 P
- DNA测序聚合酶
- 四脱氧核苷三磷酸:的dATP,dGTP,的dCTP,dTTP的
- 四脱氧三磷酸核苷(缺少2'-和3'-羟基的三磷酸核苷):的ddATP,的ddGTP,ddCTP的,ddTTP(图4)
桑格测序是一个修改形式的DNA复制。引物杂交到模板的特定位点与聚合酶结合,并结合核苷酸组装模板的反向互补拷贝。这一过程将不提供在模板上的序列和用于此目的4测序反应在分开的试管中进行的任何信息。在每个管中的关键成分的少量,2',3'-二脱氧核苷三磷酸的溶液。双脱氧三磷酸核苷的ddATP加入到管1,的ddGTP至管2的ddCTP至管3和ddTTP到管4。

图4 | 双脱氧核苷酸triphosphoates的结构(的ddNTPs)
该聚合酶不三磷酸脱氧核苷(dNTPs浓度)和双脱氧核苷三磷酸(的ddNTPs)区分,所以要么可以在每个步骤中添加。如果添加dNTP的是,DNA链将继续增长; 如果加入的ddNTP,则该DNA链将终止,因为它没有3'-羟基基团,以与传入核苷三磷酸的反应:没有进一步的核苷可以增加。在每个管的结果是不同长度的寡核苷酸,都端接一个特定的ddNTP的混合物在管1的所有终端将在A,在管2在G,在管3中C,并在管4在T该寡聚物可然后根据其大小通过电泳进行分离。如果所有四个梯子上的聚丙烯酰胺凝胶上并排,并且凝胶暴露于照相胶片,所述32 P标记的片段将产生可用于读取DNA序列(其将是反向的图像模板的补体)(图5)。在实践中有可能通过该方法测序周围的DNA的300个碱基。
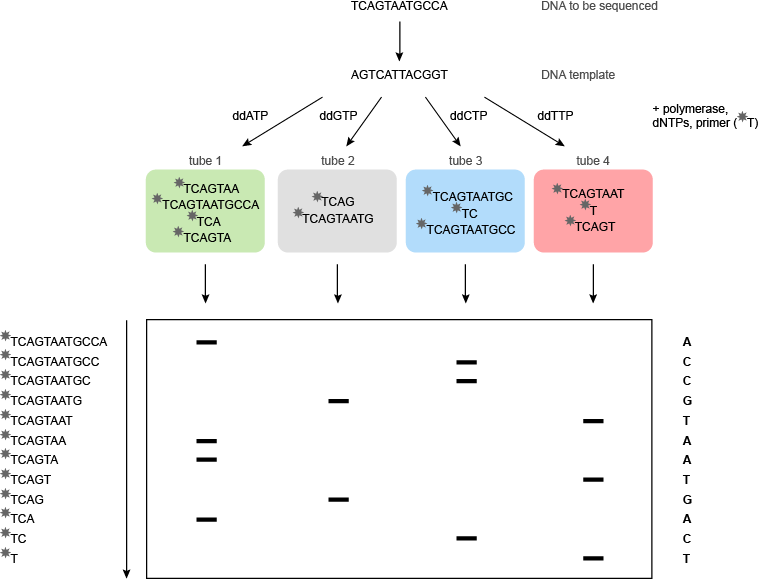
图5 | 桑格(双脱氧)测序
基于荧光二脱氧DNA测序
在Sanger测序的自动化高通量荧光版本,未标记的寡核苷酸引物使用,具有耐热性DNA聚合酶,四正常的脱氧核苷三磷酸,和具有四个双脱氧三磷酸核苷沿不同的荧光标记上它们(图6)。
现在只有1测序反应是必要的,因为终止在的ddA使DNA片段的特定原子荧光颜色,子ddG不同的颜色,DDC第三颜色和DDT的第四颜色。所述荧光染料的性质取决于所使用的DNA序列,但基本要求为四染料具有良好分辨荧光发射光谱。一个常见的系统采用FAM,JOE,TAMRA和ROX四大染料(见表1)。
表1 ⎪ 基于fluorescece测序中使用荧光染料; 它们的吸收(激发)和发射和颜色的波长
| 染料 | 最大。吸收波长/ nm的 | 最大。发射波长/ nm的 | 颜色 |
|---|---|---|---|
| FAM | 495 | 520 | 蓝色 |
| JOE | 530 | 555 | 绿色 |
| TAMRA | 550 | 575 | 黄色 |
| ROX | 580 | 605 | 红 |
的片段通过电泳分离和荧光染料由激光激发。凝胶图像然后可以由计算机进行分析和DNA序列产生。超过800个碱基可以在一个单一的凝胶泳道中读出。DNA自动测序仪可以分析在单一凝胶(从一个凝胶76 800即约基地)96个不同车道,可以分析3凝胶,每天给人每天大约有230 400个碱基或每年超过50 000 000基地的吞吐量。机已经发展到同时分析384测序反应。其他最近的创新包括标有两个荧光染料(“大染料化学”),双脱氧三磷酸核苷的发展。一种染料(通常荧光素)在其λ的激发最大值在520nm处495处的结果中发射其通过FRET转移到具有λ第二染料最大值接近520纳米。第二染料在较高波长强烈荧光。这将产生比通过第二荧光染料的直接激发在495nm处可以得到更强的荧光信号。的进步也已经在凝胶技术制成。利用毛细管凝胶,而不是平板凝胶促进了样和分析的自动化,提供更高的吞吐量。
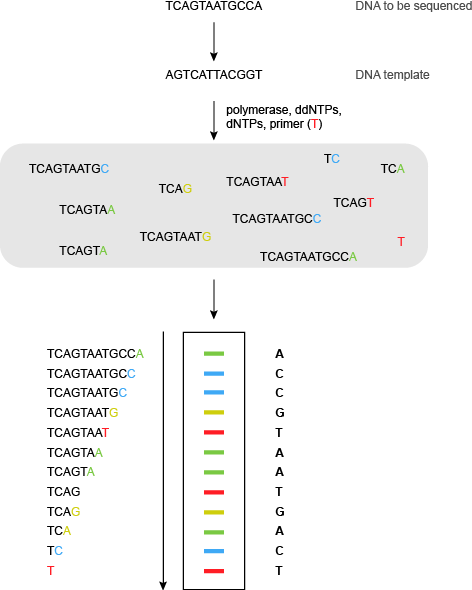
图6 | 荧光桑格(双脱氧)测序
新一代测序
人类基因组计划完成了人类基因组的3比永碱基对的测序完成,在1990年和2003年之间的巨大协同努力的人类基因组计划依靠的结果(尽管高优化和自动)Sanger测序。现在可以在几天之内,以序列的整个人类基因组,由于采用了新一代统称为下一代DNA测序测序技术的。
测序修饰的DNA
现有的DNA测序方法(包括下一代测序)是不能检测修饰的碱基。与在感兴趣的最近激增表观遗传学,未能胞嘧啶和5-甲基(这两个构成沃森-克里克碱基对与鸟嘌呤)区分是当前的测序技术的一个严重的缺点。
亚硫酸盐测序
亚硫酸氢盐(HSO 3 - )脱氨非甲基化胞嘧啶向尿嘧啶,但不与甲基胞嘧啶(反应图7)。这提供了含有5-甲基胞嘧啶碱基测序的DNA的方法。前和亚硫酸氢盐处理后的DNA进行测序:从胞嘧啶到尿嘧啶任何变化被归因于非甲基化胞嘧啶,而被假定保持亚硫酸氢盐处理后的胞嘧啶碱基的原始样品中的被甲基化。
转换非甲基化胞嘧啶向尿嘧啶,但不转换甲基胞嘧啶到胸腺嘧啶。")
图7 | 胞嘧啶重亚硫酸盐转化为尿嘧啶亚硫酸氢盐(HSO 3 - )非甲基化胞嘧啶转换为尿嘧啶,但不转换甲基胞嘧啶到胸腺嘧啶。
之前和之后的DNA的测序,其脱氨非甲基化胞嘧啶碱基为尿嘧啶,允许确定DNA样品的甲基化状态。")
图8 | 亚硫酸氢盐测序的DNA测序用亚硫酸氢盐处理之前和之后(HSO 3 - ),该脱氨非甲基化胞嘧啶碱基为尿嘧啶,允许确定DNA样品的甲基化状态。
市售重亚硫酸盐转化试剂盒使该过程例程,但它仍然是昂贵的:必须注意,所有未甲基化的胞嘧啶脱氨,并且每个DNA样品必须(当然)进行测序两次。
DNA指纹图谱
DNA指纹图谱是在上世纪80年代在英国莱斯特大学发明了亚历克·杰弗里斯爵士。人DNA可通过该方法来分析在基因水平肯定比以前的法医方法如血型判定或传统指纹分析的远较大程度来识别个人。DNA指纹(也称为DNA分析,DNA分型或基因指纹)也可用于确定个体之间的关系(例如亲子鉴定)。
DNA指纹图谱迅速成为英国国家DNA数据库,其中包含数以百万计的个人档案的技术基础。现在是在检测和罪犯定罪经常使用的主要资源,并产生数百每星期在犯罪现场发现的DNA匹配。短串联重复序列的分析序列(STR)形成在世界各地使用法医DNA分析系统的基础。
短串联重复序列
两个随机选择的人的DNA的相差在1000个碱基左右1; 换句话说,我们是99.9%相同。正是这种相似性,使得适用于所有美国的“参考”人类基因组测序。我们的DNA的一定区域包含比其他的差异,和短串联重复序列是DNA的一个区域呈现个体之间的大变化的一个例子。
DNA指纹取决于短串联重复序列(STR),两个或更多个核苷酸的短的重复模式(例如(的分析CA)ñ或(ACGT)ñ,其中Ñ是几百)。例如,序列CGTCAG在CAC,所述二核苷酸的CA重复13次(Ñ = 13)。
数千种不同的短串联重复序列,或几十微已在人类基因组中已经确定。可疑交易是在人口中的不同成员在染色体上的相同位置(基因座)观察到的,但重复的次数(Ñ个体之间)变化。在重复次数这种变化的一个例子多态性。
STR分析
STR分析使用PCR技术来测量在特定基因座的重复次数。引物结合于特定STR基因座的DNA和,通过PCR延伸。PCR产物的长度取决于重复的次数。如果PCR引物被标记,PCR产物将被标记,从而在反应结束时检测出的产物。对于每个基因座,将有两个PCR产物(每个两个等位基因)。
多个不同的STR基因座的同时分析使个人的特有配置文件被建立起来。几个PCR反应在不同的STR基因座单管同时进行,得到几种产品(两个用于每个基因座)。以下组件是必需的:
- DNA样本,例如从犯罪现场一个人的头发,或从犯罪嫌疑人的嘴凑口腔细胞
- 两种寡核苷酸PCR引物:标记在5'端用一个引物32 P,和一种未标记的反向引物
- 热稳定DNA聚合酶
- 四脱氧核苷三磷酸:的dATP,dGTP,的dCTP,dTTP的。
当标记的PCR产物在聚丙烯酰胺凝胶上运行,它们根据大小分离。结果是一个“DNA梯子”,即一个单独的(的特性图9)。
,即重复多次,二,三或四核苷酸单元中每个人的基因组中存在。 当通过PCR使用标记的引物扩增中,产生不同长度的标记的DNA片段。 这些片段通过凝胶电泳或毛细管电泳分离,得到的个体的独特的DNA“条形码”。")
图9 | DNA指纹图谱通过STR分析短串联重复序列(STR),二,三或重复几次,四核苷酸单位在每个人的基因组中存在。当通过PCR使用标记的引物扩增中,产生不同长度的标记的DNA片段。这些片段通过凝胶电泳或毛细管电泳分离,得到的个体的独特的DNA“条形码”。
使用多个位点提供了一个非常高度的确定性,在人口不两个个体将具有相同的轮廓(除非它们是同卵双胞胎)。一些当前法医系统使用10(如英国)或13(例如,美国)的STR基因座。含有PCR引物的标准STR基因座的试剂盒在市场上销售。
荧光STR分析
在STR分析的更现代的变型中,PCR引物标记有荧光染料。对于不同的STR基因座的引物标记有不同的荧光染料,增加第二维度的测定(图10)由于其迄今可能开发只具有良好分辨的光谱特性,三种不同的荧光染料的荧光染料的数量有限通常使用。

图10 | 荧光STR分析在荧光STR分析,PCR引物是荧光标记的,这导致荧光标记的PCR产物。利用不同的荧光标记的是指从不同的STR基因座始发的产品和频带可以更容易辨别。
亲子鉴定
在亲子纠纷分析的一个简化的例子(使用3个STR位点)。 在STR波段的一半来自母亲,一半来自父亲。 某些频段是从母亲和父亲都继承。 如果孩子的DNA图谱包含所有存在既不是母亲,也不是所谓的父亲的DNA图谱带,那么所谓的父亲不是孩子的亲生父亲。 如果在这个例子中涉嫌的父亲被指示支付子女抚养费?")
图11 | 在亲子STR分析争议的亲子纠纷(使用3个STR位点)使用STR(短串联重复)分析的一个简单的例子。在STR波段的一半来自母亲,一半来自父亲。某些频段是从母亲和父亲都继承。如果孩子的DNA图谱包含所有存在既不是母亲,也不是所谓的父亲的DNA图谱带,那么所谓的父亲不是孩子的亲生父亲。如果在这个例子中涉嫌的父亲被指示支付子女抚养费?
单核苷酸多态性(SNP)
突变发生在DNA为在错误的结果,DNA的复制和化学损坏。这些突变具有潜在危害的生物体,和一组DNA修复机制存在扭转他们。多态性是在一个DNA序列被建立并在人口稳定的,并且不有害的变化。有两个或更多个同样可接受的替代品,其中一个恰好是比其他人更常见。在一般情况下,如果一个变化已在人口的1%或更多的频率,它是一个多态而非突变。
单核苷酸多态性(SNPs,显剪刀)是多态性在人类基因组中最常见的类型。之一的两个特定碱基(等位基因)将发生在一个SNP位点; 例如,A将发生在人口的一些成员,而在其他-G。单核苷酸多态性发生每1000个碱基对约一次,构成本体的3×10的6在基因组中的变化,并倾向于在人口保持稳定。单核苷酸多态性发生在基因以及在控制基因表达的基因组的周围区域。
一个特定的SNP的一个等位基因上的基因的作用可能并不大 - 也许影响在一个微妙的方式所编码的蛋白质的活性 - 但即使微妙的影响可以影响易感性常见的疾病,如冠状动脉血栓形成或阿尔茨海默氏病。通过在疾病的已知患者研究大量的SNP能够确定疾病是否有遗传链路。在这种情况下,个人数字越大研究,遗传链路出现的可能性越大。一旦这样的链接被发现,在人口感染该病的任何个体的易感性可以预测的。SNP分析可能发挥在医药和诊断对未来的影响力越来越大的作用; 例如,在预测个体对药物的反应,使相应的处理,可以规定(药理学)。
在许多个SNP的两个等位基因有关于在人群中的频率相同,即如果可以发生在一特定基因座的两个碱基是A和G,一半人口将具有腺嘌呤,而另一半将具有鸟嘌呤。显然还没有进化压力有利于在这些基因座的等位基因的一个或另一个。这些种类的SNP的是在遗传分析非常有用的。相反,如果发生在一个非常低的水平的等位基因,所述SNP是作为分析工具有用要少得多,因为它会被很少遇到。因此,在DNA已经从大量个体(例如,从一组人患心脏疾病)的合并,这将是很难检测的次要等位基因的存在,即使是这一组中超过限额相对于未患有疾病的一组。
单核苷酸多态性的分析还可以提供关于物理特征的信息。这方面的一个例子是眼睛的颜色先试商用(Retinome™)。除了眼睛的颜色,单核苷酸多态性已与人类表型的其他功能,如头发和皮肤的颜色联系起来。大量的表型的SNP分析,相当于建造一个单独的“并图”的画面,因此是一个功能强大的伴奏STR分析法医调查员。在分析的SNP的挑战是等同于分析为单点突变的DNA,如基因多态性和基因突变的现象有关。
DNA诊断和突变检测
DNA诊断涉及与特定疾病相关的基因组或线粒体DNA的区域的鉴定和分析。通常该DNA序列将负责一个有缺陷的蛋白质的表达,但它也可以是控制基因表达的区域。当这种DNA序列已被确定,它的个体的DNA样品中存在(或不存在)必须确定。的突变DNA筛查取决于的灵敏,快速,准确和经济程序的发展,其中PCR扩增结合使用与适当的探针技术(实时PCR)。
基于DNA的探针的技术,可用于检测小至单个核苷酸取代,插入和缺失突变的差异。这样的点突变可以引起的遗传性疾病,如镰状细胞性贫血,囊性纤维化,苯丙酮尿症和亨廷顿氏病。遗传分析是很重要的预先和产后诊断,遗传信息也可以被用于个人的易感性预测外源性风险,如饮食或环境因素。传染病如麻疹,风疹,艾滋病毒,甲型和乙型肝炎,和由病原生物体如疾病沙门氏菌和念珠菌,可以使用DNA探针技术进行诊断。某些寡核苷酸基于探针的技术是足够的选择性密切相关的有机体/病毒区分,如1型和单纯疱疹病毒(HSV-1和HSV-2)的2变体。
实时荧光定量PCR
实时PCR是对结合的DNA的正常PCR扩增与PCR产物的同时检测,通常是在一个反应管中的PCR主题的变化。在PCR中,双链DNA的量与每个循环增加。的PCR的多次循环后,存在的DNA的量的大量增加。在实时PCR(也称为定量PCR,或定量PCR),结合于双链DNA被加入到PCR反应的试剂。作为双链DNA产生,所述试剂结合到新合成的DNA,并产生使反应实时监视的信号。同时PCR的目的是DNA的扩增,实时PCR的目的是DNA样品或反应的分析。
荧光实时PCR是PCR扩增和荧光检测的组合。在其最简单的形式中,荧光实时PCR包括使用的有机染料,只有当结合到DNA双链体是荧光的。当这样的染料是在PCR反应开始时加入发生荧光的增加作为DNA的数目双链增加,并且这是指示成功的PCR。的SYBR Green(图12)是结合双链DNA,并成为对结合(双链DNA染料配合物是荧光)的荧光的分子的一个例子。

图12 | 的SYBR Green 的SYBR Green I,结合于双链DNA的荧光分子的结构。
的SYBR绿实时PCR方法具有严重的局限性,因为它是无特异性,即无论是获得的PCR产物的性质的阳性结果。作为PCR是容易发生伪影,例如引物二聚体的形成,使用非选择性染料简单扩增并不总是非常丰富,并基于探测的方法提供更有意义的结果。
基于探针实时荧光定量PCR
当在人的诊断使用PCR它是某些产品的确切性质是重要的。在PCR产物(扩增子)键序列的鉴定可以通过加入荧光DNA探针来实现(一个短的合成寡核苷酸,其是在PCR扩增子的特定序列互补,并且不发出荧光,除非它结合到扩增子)在PCR反应。几种不同类型的探头满足这些标准,这些将在下面详细讨论。
当DNA探针在实时PCR中使用,只有在PCR扩增子中包含的互补的序列的荧光探针获得肯定信号:荧光信号是序列特异性的。例如,临床样品可以为导致囊性纤维化基因被测试如下:a探针是囊性纤维化基因(目标)的突变区域互补合成和PCR是在此存在对样品进行探测。在PCR扩增过程中的DNA分子(PCR扩增子)的数量增加而增加。如果突变存在时,探针 - 靶杂交体的浓度也增加,荧光信号生长在可预测的方式,表明突变的基因确实存在。但是,如果不产生荧光信号,这表明该患者不携带特定突变。在实际的临床应用两个突变体和野生型探针并行使用。患这种病的人,将给予从突变探头一个积极的信号,而未受影响的人将给予从野生型探针一个积极的信号。本病的携带者可能给来自两个探头一个积极的信号。
专门的设备是必要的荧光实时PCR和许多文书已设计用于此目的。它们都包括一个热循环仪来驱动PCR反应中,一个光源,以激发荧光染料(s)和一个荧光检测器,用电脑来控制仪器和处理数据一起。
在一般的“荧光”探针含有荧光染料和淬灭荧光。它们是在不存在靶核酸的非荧光因为猝灭剂从激发荧光团吸收的能量,并且该能量在较高波长作为热量耗散或辐射。还有的荧光淬灭两个独立的机制:碰撞淬火和FRET淬火。
该TaqMan方法
所述的TaqMan测定法是用于PCR产物的分析使用最广泛的实时的方法,并在SNP分析和突变检测广泛使用。TaqMan探针是由在一端用荧光团标记的寡核苷酸,例如5'-FAM(5'-荧光素),并在另一淬灭剂的荧光,例如3'-TAMRA的。在495处其吸收波长荧光的激发通常会导致荧光发射在525nm。然而,这落在TAMRA染料是在TaqMan探针接近的宽吸收光谱范围内,所以能由TAMRA染料由于荧光共振能量转移(FRET)被吸收和荧光在TAMRA的发光波长观察(585纳米),而不是在FAM的发射频率。
所述的TaqMan方法描述图13,在每一个冷却循环之前,伸展相,TaqMan探针杂交的PCR扩增子的互补序列。当Taq聚合酶聚合过程中遇到该TaqMan探针,酶的5'至3'外切酶活性将导致探针的消化。这在495纳米分离受体(TAMRA)和激发荧光体(FAM),现在带领由FAM在525纳米至荧光发射。从FAM没有能量传递给TAMRA现在可以发生,因为这两种荧光染料相距甚远。因此,当TaqMan探针已被消化的TAMRA染料不发荧光。总体而言,在525nm处的增加的荧光发射,并在585下降(nm)是指示阳性PCR反应,并且重要的是证实了正确的扩增子的存在。其他荧光染料可以在TaqMan探针可以使用,只要它们具有合适的荧光发射和吸收光谱。
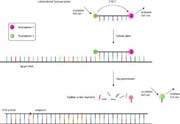
图13 | 的TaqMan分析
荧光共振能量转移(FRET)
所述的TaqMan测定法利用荧光共振能量转移(FRET),已被广泛用作光谱“尺子”,以检查的DNA二级结构的有力工具。在FRET中,激发的荧光团(能量供体)其能量通过诱导偶极-偶极相互作用时的偶极子是在大致平行的方向传送到相邻的发色团或荧光团(受体)非辐射。对于发生FRET必须有供体的发射光谱和受体的吸收光谱之间的重叠。供体和受体之间的能量转移的效率是成反比的两种荧光团(1 /之间的距离的第六功率ř 6)。的最佳距离(ř为能量的非辐射转移)为10-100埃之间为最常用的荧光团。

图14 | 荧光猝灭机制
见一节FRET淬灭剂的信息,具体的FRET淬灭剂,如BHQs。
分子信标
分子信标是一个化学修饰的寡核苷酸,采用在不存在互补靶序列的茎-环结构。该环路由一个旨在杂交PCR过程中扩增子的特定序列的探针,和杆构成的两个短的互补寡核苷酸,其中一个标用荧光团和其他与非荧光猝灭剂。在茎-环的形式中,荧光团和猝灭剂在非常靠近由短双链体结构保持,并且,如果所述荧光团被激发,能量被猝灭剂吸收,并且作为热量耗散(非辐射能量转移)。这是在分子信标(的“封闭”或“暗”状态。图15,上图)。
在PCR中的每个循环,在变性步骤中,打开的分子信标的茎环(即在干游离的碱基对或“融化”)。在随后的冷却(退火)阶段,分子信标的环路杂交在扩增子的互补序列,从而形成一个短双链体。在这种形式(“开放”的形式)的茎离得太远,联想,荧光团和淬灭剂保持分开,并且照射产生的荧光信号(图15,下图)。在实时PCR,荧光必须在在该信标中的茎环形式是稳定的(通常约50℃)的温度下进行测量,以使未杂交探针不产生不希望的背景荧光。
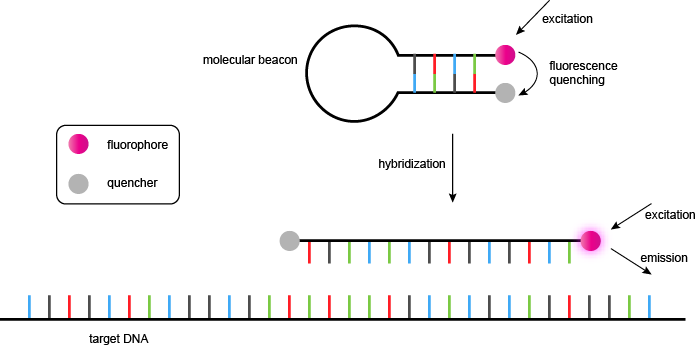
图15 | 分子信标
最初,分子信标被设计用在5'末端的荧光团5-(2'-氨基乙基) -氨基萘-1-磺酸(EDANS)和荧光猝灭剂的4-(4'- dimethylaminophenylazo)苯甲酸(DABCYL)在3'末端(图16)。

图16 | DABCYL和EDANS 荧光淬灭DABCYL和荧光EDANS的结构。
最近,更广泛的荧光团已用相同DABCYL猝灭一起使用。碰撞荧光淬灭是在封闭的形式非常有效的,并且不上淬灭剂的吸收光谱强烈依赖。然而,荧光的开放形式的水平是有限的荧光团和猝灭剂保持相对彼此接近。
分子信标可以用来通过测量荧光的强度在每个PCR循环(定量PCR)退火阶段量化扩增子的PCR过程中的浓度。他们也已经在SNP和突变分析广泛使用。几个分子信标,每一个独特的荧光团,可以在多重PCR反应中用于同时分析在不同基因座的单核苷酸多态性。
蝎子引物
蝎子是PCR引物附着(通过接头)一个分子信标充当PCR塞子。这个连接体,通常六乙二醇,隔离底漆的信标部分。蝎子的引物部分的PCR延伸后,将所得的扩增子中包含的序列是探针部分互补。在PCR循环的变性阶段扩增子呈现单链和冷却所述探针元件结合到这种补体,以形成分子内双链体。在这种形式的猝灭剂不再位于接近荧光团和荧光信号产生(图17)。
图17 | 蝎子引物
蝎子探针含有在单个寡核苷酸几种化学修饰,因此是相对复杂的分子来合成。然而,它们具有比分子信标一些主要的优点。PCR过程中的活性蝎的形成是一个分子内的过程,因此,比使用分子信标发生等效的分子间反应快得多。此外,蝎子的活性形式是比分子信标,这往往脱落他们的目标,并折叠成非荧光分子内发夹环的动力学更加稳定。在对比TaqMan探针,蝎子不取决于酶裂解以产生荧光信号,和高速PCR循环因此可能的,从而导致非常快速而可靠的检测系统。
其它诊断方法
DNA微阵列
寡核苷酸可以化学附着到材料如玻璃或硅的表面,在其上它们形成小的大约100微米的“点”(10 -4直径米)。寡核苷酸的大量可在单个幻灯片中加以规定,以形成微阵列,和荧光标记的DNA(标记的PCR产物或cDNA)的单链可通过杂交捕获。(基因是单链DNA,以从它是由合成的RNA互补的反转录,它给出了在细胞(表达分析表达的各种RNA消息)的性质间接信息)。如果这样的微阵列包含1000点则在理论上是可能的杂交一个唯一的互补核酸序列的每个点。的DNA序列的同一性,从光点的位置与它杂交使用荧光扫描仪推断。
附连到捕获的核酸链的荧光标记物可以用许多不同的方法加入。PCR产物可以在5'末端简单地通过使用含有5'-荧光染料PCR引物进行标记。PCR引物可以与多个荧光团进行标记,但是这些往往以猝灭彼此并且还抑制了PCR反应。一种更好的方式来介绍多个标签到PCR产物是在PCR使用荧光标记的脱氧核苷三磷酸或逆转录酶反应(例如荧光素标记的dT)。然而,在PCR反应的效率可以通过该杂环基,其可以抑制Taq聚合酶上的化学修饰受到损害。一个精心确定未标记和标记的脱氧核苷三磷酸的混合物,因此必须使用,并且这是罕见实现标记的密度大于每30个核苷酸1荧光更大。微阵列测定,也可以由个别的PCR产物附着到滑动作为离散斑点,并与荧光标记的寡核苷酸(池探测在反向格式进行图18)。

图18 | DNA芯片
因为DNA链的非常大的数字,可连接到单个阵列DNA微阵列是高通量突变,SNP和基因表达分析是有用的。微阵列是由适合机器人系统自动化,允许非常高的吞吐量。然而,他们目前由于对表面分子的一些不良化学和生物物理特性的挑战。首先,它难以形成非常密集阵列。100μm的点尺寸是可以实现的,但较小的斑点(例如1微米)将允许每个阵列的斑点的高得多的号码,允许使用溶液相DNA和更大的吞吐量的较小体积的。其次,互补DNA分子的一个表面上的杂交是几乎没有溶液杂交效率高。为了使系统可行,表面的性质和表面和附着的DNA之间的连接体的性质,必须仔细控制。
荧光原位杂交(FISH)
原位杂交(ISH),其允许特定的DNA序列的使用放射性标记染色体的识别和可视化,在讨论的合成和化学修饰的寡核苷酸的应用。荧光原位杂交(FISH),其中通过采用基于荧光的检测和可视化通过荧光显微镜延伸ISH,是在遗传分析的重要工具。FISH的原理在于标记探针的退火以固定的细胞或组织的染色体内的互补链,随后通过检测荧光标记的。探针(DNA或RNA)通常由三个聚合酶基于酶的方法中的一种(切口平移,随机引物或PCR),其允许荧光标记的脱氧核苷三磷酸的掺入制备。每30个核苷酸一种荧光标记的平均掺入水平是典型的。的DNA探针的长度可以是100 bp和1000bp的间。较长的探针增加的非特异性背景荧光,但短探针是很困难的,由于杂交不足和标签低水平来检测。重要的是目标为在探头访问,必须在原位,而不是由核酸酶降解被保留。可视化限制从整个染色体为40 kb的染色体区段跨越。
荧光原位杂交已经用于识别染色体内的基因的位置(虽然这是与人类基因组的测序不太有用),染色体“画”,用于可视化整个染色体的技术,并在表征和诊断疾病。

 吴医师
吴医师